
Renal failure is a condition in which the kidneys lose the ability to maintain homeostasis (removal of metabolic waste, regulation of fluid and electrolyte balance, maintenance of acid-base equilibrium, and hormonal function). In clinical practice, a distinction is made between acute kidney injury (AKI), where dysfunction develops within hours to days, and chronic kidney disease (CKD), where loss of function progresses over months to years. Both forms have their own causes, mechanisms, and stages of complications.
Causes of AKI are usually divided into three main groups: prerenal (related to reduced kidney perfusion), intrarenal (tubulointerstitial and glomerular lesions), and postrenal (obstructive).
-
Prerenal causes include severe hypovolemia (blood loss, dehydration), low cardiac output state (heart failure, cardiogenic shock), severe hypotension, or vascular disorders. Without correction of hypoperfusion, the prerenal form can progress to necrotic tubular damage.
-
Intrarenal causes are mainly acute tubular necrosis (ischemic or toxic), acute interstitial nephritis (allergic, drug-induced), acute glomerulonephritis, and vascular lesions. Risk factors include sepsis and nephrotoxins (contrast agents, cytotoxic drugs, heavy medications).
-
Postrenal AKI arises from obstruction at the ureter or lower level (stones, tumors, strictures, prostatic adenoma) and may be reversible if the obstruction is removed in time.
The mechanisms and staging criteria for AKI are described in international KDIGO guidelines (stages 1–3 by creatinine and urine output), which are crucial for prognosis and therapeutic strategy.
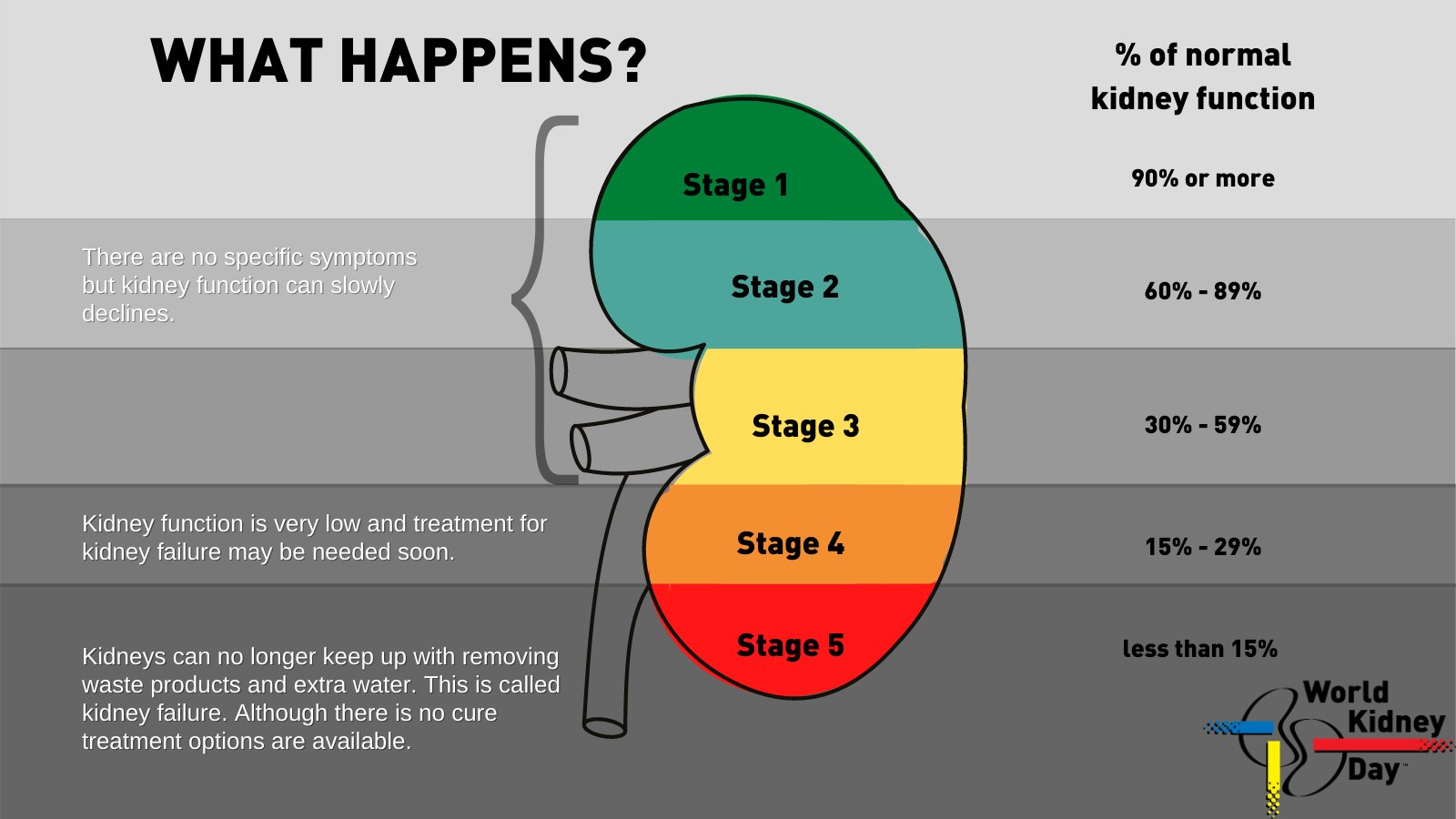
Chronic kidney disease (CKD) develops with long-term renal tissue damage and gradual nephron loss. The leading global causes of CKD are diabetes mellitus and arterial hypertension; other frequent causes include chronic glomerulonephritis, obstructive uropathies (long-term obstruction), hereditary diseases (polycystic kidney disease), chronic infections, and autoimmune disorders. Mechanistically, nephron loss leads to compensatory hyperfiltration of remaining nephrons, increased intraglomerular load, and altered hemodynamics, which further aggravates damage and stimulates progressive interstitial fibrosis and glomerulosclerosis. Clinically, CKD is classified by glomerular filtration rate (GFR) in categories G1–G5 and by degree of albuminuria A1–A3; the combination of these parameters provides prognosis and guides therapy.
The pathophysiology of progression from acute to chronic and mechanisms of CKD advancement involve multiple interconnected processes: recurrent ischemic/toxic injuries and sustained inflammation cause tubular cell death, loss of peritubular capillaries, activation of fibrogenesis, and extracellular matrix remodeling. Chronic inflammation, proteinuria, uncontrolled hypertension, and metabolic disturbances (e.g., hyperglycemia) further contribute to nephron loss. The more active the proteinuria and the lower the baseline GFR, the higher the risk of rapid progression to end-stage renal disease.
Clinical stages and complications of AKI and CKD differ by timing and severity. In AKI, the most dangerous early threats are: rising hyperkalemia with arrhythmia risk, severe metabolic acidosis, fluid overload and pulmonary edema, uremia with impaired consciousness, and in infectious-toxic scenarios—multiorgan dysfunction. If AKI does not resolve, it may progress to CKD. The severity of AKI (KDIGO 1–3) correlates with mortality risk and likelihood of later CKD.
In CKD, as GFR declines, systemic complications accumulate. In early stages, symptoms may be minimal but subtle metabolic changes already occur. With progression, marked uremia (nausea, appetite loss, neuropsychiatric disturbances), anemia (due to erythropoietin deficiency and iron metabolism disorders), mineral-bone disorders and secondary hyperparathyroidism (CKD-MBD) with fracture risk, hypertension, hyperkalemia, metabolic acidosis, immune suppression (leading to infections), malnutrition, and cachexia appear. Cardiovascular complications are the leading cause of death in CKD patients: uremic toxins, hypertension, electrolyte imbalance, and T-cell/inflammatory alterations accelerate atherosclerosis, heart failure, and arrhythmias.
Main factors accelerating progression and impairing renal regeneration include persistent proteinuria, uncontrolled hypertension, poor glycemic control in diabetes, recurrent AKI episodes, smoking, obesity, older age, and comorbidities (especially cardiovascular and vascular disease). Slowly but continuously, chronic interstitial hypoxia, persistent inflammation, and active fibrosis prevent tissue recovery and promote irreversible scarring.
Monitoring and markers include GFR estimation (eGFR by creatinine), serum creatinine and urea dynamics, quantitative assessment of albuminuria/proteinuria (albumin-to-creatinine ratio), electrolyte monitoring (calcium, phosphate, potassium), hemoglobin and mineral metabolism markers, as well as blood pressure. Prevention and slowing of progression rely on early identification of reversible causes (volume/perfusion correction, obstruction relief, nephrotoxin withdrawal), strict risk factor control (glycemia, blood pressure, dietary protein if needed), and preparation for renal replacement therapy (dialysis or transplantation) at the terminal stage.
Summary: Causes of renal failure are diverse and include vascular, metabolic, inflammatory, and obstructive factors. AKI develops rapidly and poses immediate life-threatening electrolyte and fluid disturbances, whereas CKD progresses more slowly but manifests with multiple systemic complications (anemia, CKD-MBD, cardiovascular disease, infections, etc.).
Renal Tubule Epithelial Cells (RTEC) in Therapy
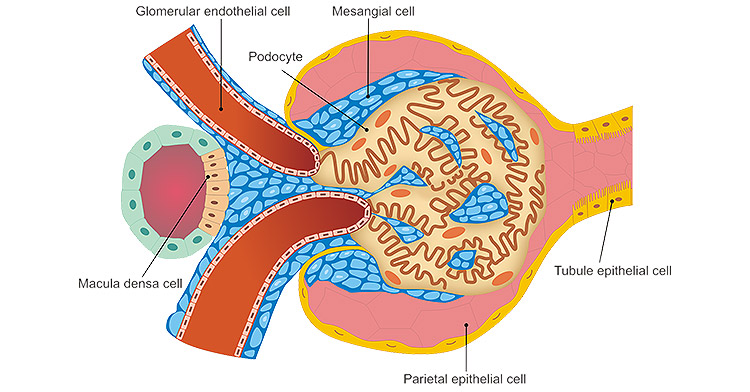
Renal tubular epithelial cells (RTEC) are increasingly studied as a promising tool for renal failure treatment, especially in the context of AKI and early CKD. Their application is linked to the critical role of tubular epithelium in homeostasis and nephron regeneration.
Why tubular cells?
The tubular epithelium not only performs filtration and reabsorption but also serves as a source of signaling molecules regulating inflammation, angiogenesis, and interstitial remodeling. In kidney injury, proximal tubular cells are most often affected by ischemia and toxins, becoming the trigger point for necrosis, apoptosis, and inflammatory response. Thus, restoring or replacing these cells is crucial.
Mechanisms of therapeutic action of RTEC:
-
Direct regeneration: Transplanted cells integrate into damaged nephrons, restoring morphology and partial function.
-
Secretory/paracrine activity: RTEC release growth factors (VEGF, HGF, EGF), cytokines, and exosomes that stimulate endogenous renal stem cells and suppress fibrogenesis.
-
Immunomodulation: Secreted molecules reduce activity of inflammatory macrophages and T-cells, decreasing chronic injury.
-
Anti-apoptotic/antioxidant effect: Transplanted RTEC or their exosomes reduce oxidative stress and apoptosis in host tubular cells.
Strategies in use:
-
Direct transplantation (systemic or local), improving creatinine/urea and tubular morphology.
-
Bioartificial kidney devices (renal assist device), where RTEC-based cartridges perform metabolic functions during dialysis.
-
Exosome-based therapies, focusing on cell secretome as a safer, standardized product.
-
Genetically modified RTEC to improve survival or enhance protective factor production.
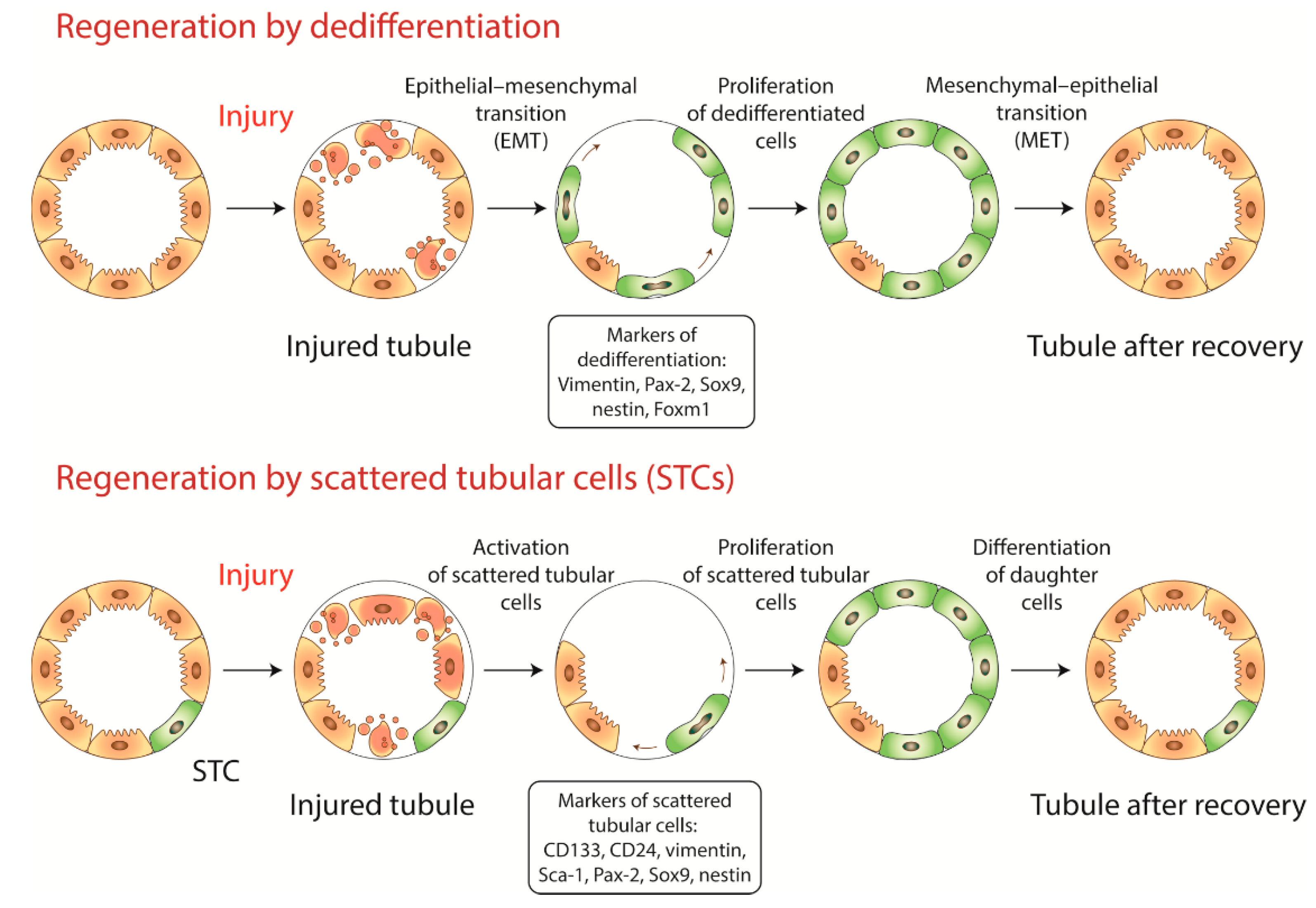
Integration process of RTEC into damaged kidney:
-
Migration and homing: Guided by chemokines (SDF-1, CXCL12, HGF), RTEC reach the injury site. Local delivery increases targeting precision.
-
Adhesion: Cells attach to extracellular matrix of injured tubules via integrins and adhesion molecules (ICAM-1, VCAM-1).
-
Integration: Cells incorporate into tubular epithelium, polarize, form brush border (proximal tubules), and tight junctions.
-
Functional activation: Restored cells resume transport of ions, glucose, amino acids, and acid-base regulation. Markers such as Na⁺/K⁺-ATPase, AQP1, and glucose transporters reappear.
Cell survival:
-
Currently limited: many transplanted RTEC survive only 1–3 weeks in tissue.
-
Causes: ischemia, poor vascularization, inflammatory microenvironment.
-
Strategies: hydrogels, 3D scaffolds, hypoxic preconditioning, MSC co-administration to support angiogenesis and survival.
-
Importantly, even short-lived cells release exosomes and signals that activate endogenous repair, often producing therapeutic benefit beyond actual survival.
Expected effects:
-
Short-term (days–weeks): Reduced inflammation, decreased necrosis/apoptosis, improved renal clearance (lower creatinine/urea).
-
Mid-term (weeks–months): Restored tubular morphology, improved cortical microcirculation, reduced fibrosis via TGF-β suppression.
-
Long-term (months–years, still limited clinical data): Slowed CKD progression, reduced transition from AKI to fibrosis, prolonged nephron function, potentially delaying dialysis or transplantation.
In conclusion: RTEC can integrate into damaged tubules and partially replace dead cells, but their main contribution lies in secreted factors that stimulate regeneration and protect endogenous cells. Survival remains limited, so future strategies focus on combining RTEC with biomaterials or shifting toward exosome-based therapies as a more stable, standardized approach.